On Friday (November 1st), I did a lot of work with analyzing peptide mapping data using the Origin program. But before I get into that, JP said that the peptide formation that I prepped for last week came out perfect! Anyway, I worked with three different sets of data from the same experiment- one set of 20 peptides and two sets of 56. After copying the data from a previous Excel sheet, my first step was to change the settings of all of the odd-numbered columns to "Y-error" because it represented standard deviation data. After I had done that for all three sets of data, I adjusted the fit curves by using a one-site bind pharmacology non-linear cure fit. In that process, I had to change the k-values of every data set to 1 to standardize the fits.

Our goal is to look at the data for each of the peptides and determine which peptides have low Kd (affinity towards the substrate) values and high R-squared values (good fit to the best-fit line). The area we are looking for is represented by the green area above. In looking at the plots, I narrowed the range to an R-squared value of 0.5 to 1 and Kd value of 3E-6 to 8E-6, and changed the data point labels to display their peptide numbers. Looking at the layout of peptides in this region, we determined that the most accurate set of data is the second set of 56, which makes sense because those peptides were made in 2012, while the peptides used in the other two sets were made in 2011.
After looking at those plots, I the plotted Peptide # vs. 1/Kd. This produced plots with many sporadic troughs and peaks. JP introduced me to the idea of curve-smoothing. Using the Kd values of the peptides, we are ideally looking for the Kd values of individual amino acids. To do this, we use averages of the Kd values of different peptides. For example, A would equal KD1, B would equal the average of KD1 and KD2, etc.
Using an Excel template that JP made for a previous experiment, I plotted averages of three up to fifteen peptides. While the fifteen-peptide averages obviously smoothed the curve the most, it is hard to determine the correct plot we should use because as the curve gets smoother, it hides certain important peaks.
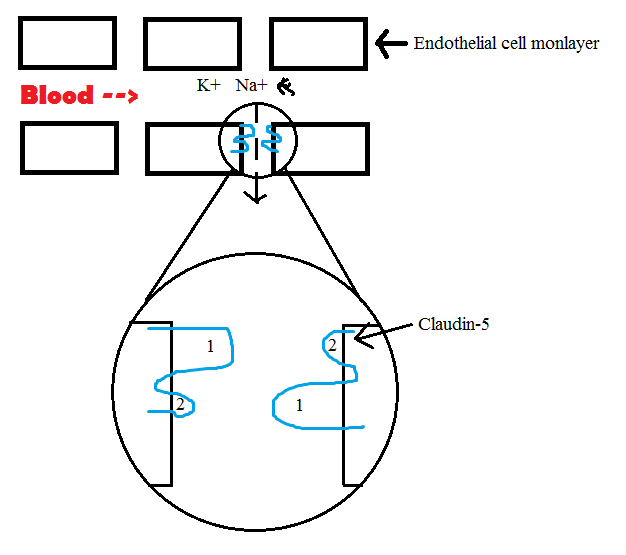
After I finished my analysis work, JP showed me some of the brain cells that he is culturing! He just started growing them on Tuesday, so they had not yet grown into a full layer of cells. They will be used to run a second trial of an
experiment we did last winter, which involves testing the permeability of the blood-brain barrier by passing different sizes of sugar molecules through a layer of cells. I can't wait to continue working with the data and see where that experiment goes!






.png)






